Databases
An introduction to using databases for bioinformatics
Jer-Ming Chia (chia@cshl.edu)
What we will do today
- Creating databases, tables in MySQL
- Querying and manipulating data using SQL
- Querying and manipulating data using Perl DBI
What is a database
- A collection of data
- Text file with a list of genes
- GFF text file
- BAM file
- Excel spreadsheet
- Set of tables in MySQL
A table of genes

- Each row is a record of a gene
- Each column is a set of values constrained by a type
- A simple query:
What is the location of gene ‘GRMZM2G42775’?
A more complex query
Gene table
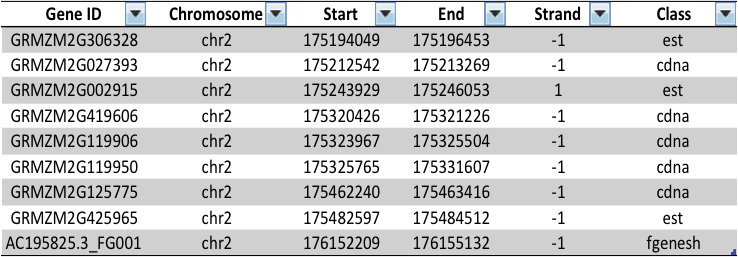
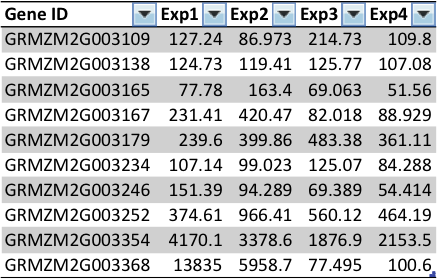
Expression data
Gene Ontology

"What are the classes of highly expressed genes in region 50Mb-55Mb of chromosome 5?"
DBMS: Software for managing databases
- Database Management Systems (DBMS)
- General term for software for managing data
- Creating tables
- Loading data
- Querying data
- E.g:
MySQL, SQLite, Oracle, Microsoft Access, Berkley DB, MongoDB RDBMS Relational Database Management System- Software for managing related data that is stored across multuple tables
Using a DBMS
Through a user-interface
- E.g: MySQL workbench, HeidiSQL, SequelPro, SQLite Manager, SQLite Spy
Programmatically through SQL
Structured Query Language
E.g: “select gene_id from gene_table where chromosome = ‘chr2’;”
Programmatically through an API in another programming language
Perl DBI
Java JDBC, C ODBC
MySQL
- A robust RDBMS that is very popular for large bioinformatics databases. Great for:
- Very large, persistent datasets
- Multi users with different permission levels
- High volume transactions
- To access the mysql client from the command line, you need 4 pieces of information:
- Host ( Defaults to localhost)
- Port (Defaults to 3306)
- Username
- Password
Using MySQL Shell
Start the MySQL client from the command line, and this will bring up a MySQL shell, connected to the MySQL server on you local machine:
$ mysql –u root
To connect to a remote server, for example the public Ensembl MySQL:
$ mysql –h ensembldb.ensembl.org –P 5306 –u anonymous
In the MySQL shell, each line of commands must terminate with a ‘;’
mysql> show databases;
mysql> create database myTestDB; # myTestDB is the database name
mysql> use myTestDB; # Use database myTestDB
mysql> help;
To quit:
mysql> \q;To cancel a command:
mysql> \c;Creating a table
- Things to consider:
- Table name
- Name of each column
- Data type of each column
- Range of values of data in each column
Basic Datatypes
- Numeric
- INT : for integers
- Double : numerical data with decimals
- Strings
- CHAR : for strings up to 255 in length
- TEXT : large strings
- Lots more on the MySQL website http://dev.mysql.com/doc/refman/5.6/en/data-types.html
- And also, a cheat sheet of course. http://en.wikibooks.org/wiki/MySQL/CheatSheet
SQL: Creating a table
Syntax:CREATE TABLE tablename ( column_1_name datatype [optional constraint] column_2_name datatype [optional constraint] ... );
For example, to create this table using SQL:
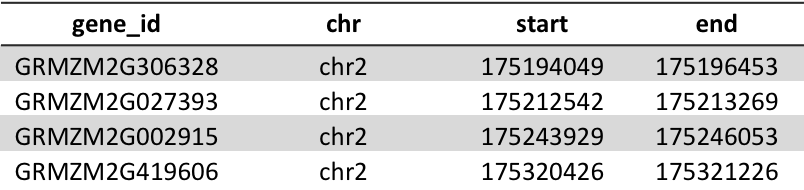
CREATE TABLE genes (
`gene_id` char(25) NOT NULL DEFAULT '',
`chr` char(5) NOT NULL DEFAULT '',
`start` int(9) NOT NULL DEFAULT '0',
`end` int(9) NOT NULL DEFAULT '0',
PRIMARY KEY (`gene_id`)
);
Keys and Indexes
- INDEX
- Synonymous with KEY
- It is the lookup column, or a set of columns, for a table.
- There can be more than one KEY in a table
- PRIMARY KEY
- The primary key for a table represents the column, or set of columns, that is mostly frequently used as an index to the table
- Columns used as the primary keys must be contain values that are unique to each row
- There can only be one primary key in a table
SQL : Simple Queries
- Query using SELECT
- Use LIMIT when the list is too long
- Wildcard character “*” for all columns in table
mysql> use progbio2012; # Use database progbio2012
mysql> SELECT gene_id from genes; #SELECT Column [columns] from Table
SELECT gene_id, chr, start, end FROM genes limit 10;
SELECT * from genes limit 10;
SQL: SELECT ... WHERE for filtering results
SELECT gene_id FROM genes WHERE chr=‘chr5’;
# what are the genes that lie within 50Mb – 55 Mb of chromosome 5?
SELECT gene_id FROM genes
WHERE chr=‘chr5’ and end >= 50000000 and start <= 55000000;
SQL: SELECT ... WHERE with OR
SELECT gene_id , class
FROM genes
WHERE chr=‘chr5’
and (evidence=‘cdna’ OR evidence= ‘est’);
SQL: Sorting and Distinct
SELECT gene_id, chr, start, end FROM genes
WHERE chr=‘chr10’
ORDER BY end desc
LIMIT 20;
# What is the unique list of gene evidences in the genes table?
SELECT DISTINCT evidence from genes;
SQL: SELECT COUNT… GROUP BY
SELECT COUNT(*) FROM genes;
# How many genes lie in chromosome 5?
SELECT COUNT(*) From genes where chr=‘chr5’;
# How many genes are there in each chromosome?
SELECT chr, COUNT(*) FROM genes GROUP BY chr;
Other SQL functions besides COUNT:
- avg, min, max, concat
SQL: Select ... Join
Gene table
Expression data
Gene Ontology
SQL :Simple JOIN
SELECT genes_go.gene_id, go_id, day1
FROM genes_go, expression
WHERE genes_go.gene_id = expression.gene_id
and go_id = 'GO:0030528';
Let’s try this
“What are the classes of highly expressed genes in region 50Mb–55Mb of chromosome 5?"
Perl DBI
DBI is a module that provides access to DBMS in Perl
It hides the nuts and bolts for connecting to each type of DBMS, leaving a consistent interface for connecting to a database
The key object in DBI is the database handle ($dbh), which represents a connection to a DBMS.
Perl DBI: 3 easy steps
Create a database handle
$dbh = DBI->connect(....) Execute a SQL statement using the database handle
$dbh->do something Disconnect the handle
$dbh->disconnect
Perl DBI Step 1: Constructing the handle
- for MySQL:
my $dbname = "progbio2012"; my $driver = ‘mysql’; my $user = 'root’; my $passwd = ''; my $host = 'localhost'; my $port = 3306; my $dsn = "DBI:$driver:database=$dbname;host=$host;port=$port"; my $dbh = DBI->connect($dsn,$user,$passwd); - for SQL lite:
my $dbname = "prog2012"; my $driver=‘SQLite’; my $dsn = "DBI:$driver:$dbname"; my $dbh = DBI->connect($dsn);
Perl DBI Step 2: Executing the SQL
- Construct the SQL query
my $sql = “SELECT count(*) From gene”; - Execute the transaction using the database handle
Querying and fetching data: my $results_array_ref = $dbh->selectall_arrayref($sql);
DBI Example: Executing an SQL
For example, to create a table using
=pseudocode $dbh = DBI->connect($dsn) $dbh->do(SQL) $dbh->disconnect =end
#! /usr/bin/perl
use strict;
use warnings;
use DBI;
my $dbname = ‘progbio2012’;
my $user = ‘root’;
my $passwd = ‘’;
my $host = ‘localhost’;
my $port = 3306;
my $dsn = "DBI:mysql:database=$dbname;host=$host;port=$port";
my $dbh = DBI->connect($dsn,$user,$passwd);
my $sql = "CREATE table foo (bar char(10));" ;
$dbh->do($sql);
$dbh->disconnect;
exit;
DBI : Fetch a list of genes using DBI
=pseudocode $dbh = DBI->connect($dsn) $fetched_results = $dbh->fetch SQL query do something with results $dbh->disconnect =end
#! /usr/bin/perl
use strict;
use warnings;
use DBI;
my $dbname = 'progbio2012';
my $user = 'root';
my $passwd = '';
my $host = 'localhost';
my $port = 3306;
my $dsn = "DBI:mysql:database=$dbname;host=$host;port=$port";
my $dbh = DBI->connect($dsn,$user,$passwd);
my $query = "SELECT gene_id, chr, start, end from genes";
my @results = @{$dbh->selectall_arrayref($query)};
foreach my $row_ref ( @results){
my $str = join "\t",@{$row_ref};
print $str,"\n";
}
$dbh->disconnect;
exit;
DBI : Count using selectrow_arrayref
For SQL queries which will only return a single row,
e.g: SELECT COUNT query
=pseudocode $dbh = DBI->connect($dsn) $fetched_row = $dbh->fetch SQL query do something with results $dbh->disconnect =end
#! /usr/bin/perl
use strict;
use warnings;
use DBI;
my $dbname = 'progbio2012';
my $user = 'root';
my $passwd = '';
my $host = 'localhost';
my $port = 3306;
my $dsn = "DBI:mysql:database=$dbname;host=$host;port=$port";
my $dbh = DBI->connect($dsn,$user,$passwd);
my $query = "SELECT COUNT(*) from genes where chr ='chr10'";
my @row = @{$dbh->selectrow_arrayref($query)};
print join ("\t",@row),"\n";
$dbh->disconnect;
exit;
DBI : Querying using placeholders
Fetch the expression level for a list of genes
=pseudocode $dbh = DBI->connect($dsn) $sth = $dbh->prepare(SQL) loop: $sth->execute $sth->fetchrow_array; end loop $dbh->disconnect =end
my $dbh = DBI->connect( $dsn, $user, $passwd );
my $query = "SELECT day1, day2, day3, day4
FROM expression
WHERE gene_id = ?";
my $sth = $dbh->prepare($query);
my $file =shift;
open IN,"<",$file || die ("Can't open file $file $!");
while (my $gene_id = <IN>){
chomp $gene_id;
$sth->execute($gene_id);
my @results = @{ $sth->fetchall_arrayref};
foreach my $row_ref (@results) {
my $str = join "\t", @{$row_ref};
print $gene_id,"\t",$str, "\n";
}
}
close IN;
$dbh->disconnect;
exit;
DBI: Placeholders vs SelectAll
# fetch expression level for a list of genesPlaceholders:
=pseudocode @genelist = read from file $dbh = DBI->connect($dsn) $sql = “SELECT day1 FROM expression WHERE gene_id = ?” $sth = $dbh->prepare($sql) loop through genelist: $sth->execute($gene) $expr = $sth->fetchrow_array print $expr end loop $dbh->disconnect =end
- 1 database transaction for each gene queried
- Slow if you have many genes to query
SelectAll:
=pseudocode
@genelist = read from file
$dbh = DBI->connect($dsn)
$sql = “SELECT gene, day1
FROM expression”;
%gene_expr hash
for each result in
$dbh->selectall_arrayref($sql):
$gene_expr hash{$gene} = $expr
end for each result
loop through genelist:
if exist in %gene_expr hash
print $expr
end loop
$dbh->disconnect
=end
- A single database transaction
- Slow if you’re fetching millions of rows and you end up only needing a small fraction
For other DBI functions, see CPAN
With $dbh->selectall_hashref
my $dsn = "DBI:mysql:database=$dbname;host=$host;port=$port";
my $dbh = DBI->connect($dsn,$user,$passwd);
my $query = "SELECT gene_id, chr, start, end from genes";
my %results = %{$dbh->selectall_hashref($query,'gene_id')};
foreach my $gene (keys %results){
print $gene,"\t";
$results{$gene}->{'chr'},"\t",
$results{$gene}->{'start'},"\t",
$results{$gene}->{'end'},"\n";
}
Other useful MySQL commands
For loading a text file into a table from the unix command line:
$ mysqlimport --local –u root databasename filename.txt
For dumping data from a table:
$ mysqldump –u root databasename tablename > table.sql
Or for dumping the entire MySQL database:
$ mysqldump –u root databasename > database.sql
Other useful SQL commands
Inserting new rows into table
INSERT into genes (gene_id, chr, start, end, evidence)
values (‘foo’,’chrX’,1000,1500,’cdna’);
Updating data in table
UPDATE genes SET gene_id = ‘bar’ WHERE gene_id = ‘foo’;
Before you leap:
- What we did not cover and should be considered before charging ahead and designing your own databases
- The full JOIN Syntax:
-
mysql> SELECT genes_go.gene_id, go_id, day1 from genes_go join ( expression ) on ( genes_go.gene_id = expression.gene_id ) where go_id = 'GO:0030528';
- Database Normalization
- Consider other RDMBS: e.g SQLite
Problem sets: Databases and Perl DBI
- For this problem set, we have installed MySQL servers in each of the machines.
- In each MySQL server, you'll find a database named progbio2012.
- This database has 5 tables: genes, genes_go, expression, snp, go_terms
- 'genes' table lists the location and evidence class of each gene
- 'genes_go' table contains the Gene Ontology terms for genes that have a Gene Ontology annotation
- 'go_terms' list the Gene Ontology descriptions of the GO Ids
- 'expression' table contains the expression level of genes in 4 experiments.
- 'snps' list of SNPs in chr1 and chr10.
- You will need the data in these tables for the problem sets
Problem sets: Getting familiar with mySQL
- Follow the steps below to enter the MySQL shell and use the database progbio2012.
on the unix command line:
You're now in the MySQL shell.$ mysql –u root To use the database progbio2012:
mysql> use progbio2012;
mysql> show tables;
Problem sets: Getting familiar with mySQL
- Use the 'explain' command to see the schema of each table
on the unix command line:
mysql> explain genes;Try out SHOW command to see the SQL-CREATE syntax for each table:
mysql> show create table genes;
Problem sets: SQL
- Using SQL, perform the following queries:
- How many rows are there in the gene table?
- How many genes have GO annotations?
HINT: count(distinct gene_id) - List the number of genes in each evidence class in the genes table
HINT: Using a COUNT ... GROUP BY query - Using a SQL query that joins the genes_go and expression table,
select the day1 value of genes that have the go_term 'chromatin binding' ('GO:0003682') - Try the query above again, but limit it to genes on chr1.
Problem sets: Perl DBI
- Do the following using Perl-DBI
- Using a similar query to Q3.d above, write a Perl DBI script that produces a tab-delimited text file for genes with go_term 'nucleic acid binding'. (GO:0003676)
The text file should have the columns: gene_id, go_term, day1, day2, day3, day4 - Construct a query to find genes where the expression level in day4 is greater than day1.
Print out this list of genes.
- Using a similar query to Q3.d above, write a Perl DBI script that produces a tab-delimited text file for genes with go_term 'nucleic acid binding'. (GO:0003676)
- Advanced problems
- How many genes in the first 100Mb of chr1 contain snps?
- Compute the gene density on chr10 across 1Mb windows.
Assume that Chr10 has a total length of 150Mb. - Compute the average expression level in each experiment (day1, day2, day3, day4) for the genes in each GO term.